Lots of people have done phage therapy, but in most cases, phages and bacteria haven’t been tracked over the course of therapy. So while we might know if the patient got better in the end, we don’t really know what happened in the body. We don’t know how many phages stick around in the system, or for how long. We don’t know how long it takes to get rid of the target bacteria. We don’t know if it looks like an arms race, with phage and host taking turns going up and down in numbers, until one wins (hopefully the phage!). Or maybe the phage kills all the bacteria after the first dose, and there’s no need for further doses? All these things and more would be super interesting to know.
qPCR: a way to count phages and bacteria
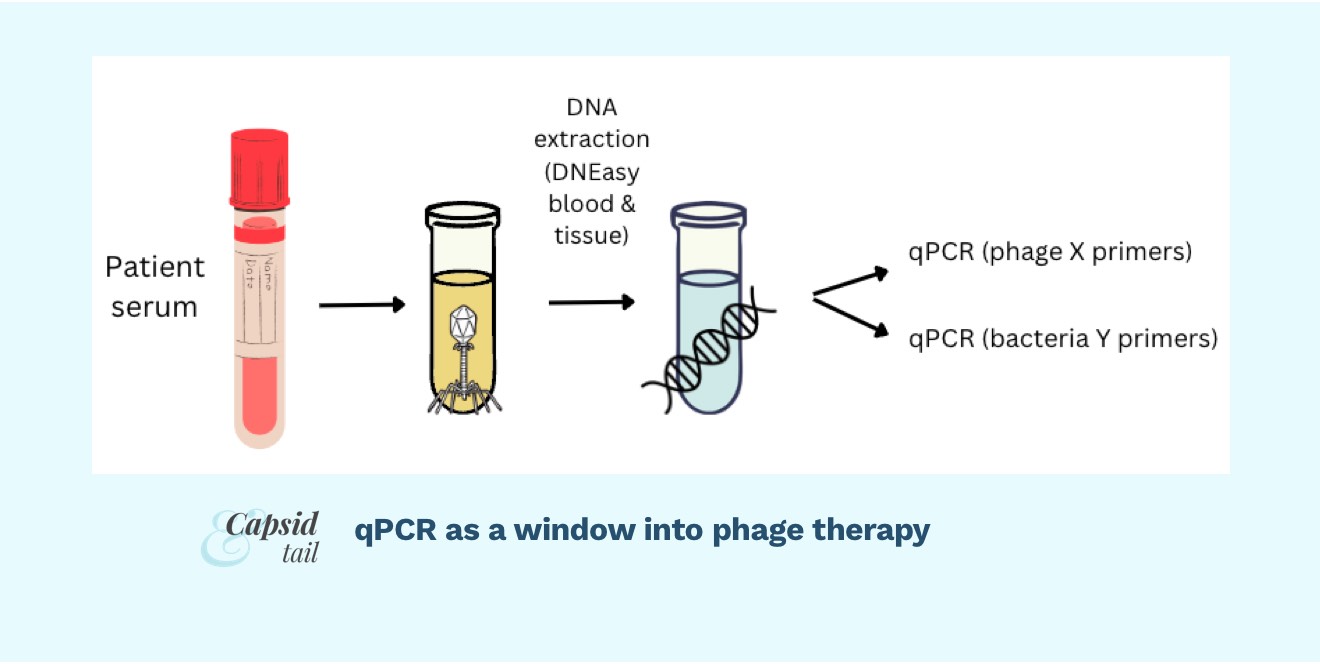
One way to “watch” the phage-bacteria fight is using qPCR to track each phage and each bacteria over the course of treatment. To do this, you could take blood samples, extract DNA, and use PCR to amplify a gene from your phage, and a gene from your target bacteria, and in essence “count” the number of each in the patient’s blood. You could take blood samples over time, say once a day during a course of phage treatment, and then graph your phage and bacterial numbers over time.
⛔ Major caveat: qPCR alone is not enough to say what’s going on with your phage or bacteria… it is useful in combination with other methods, though! One key reason is that qPCR only detects DNA, but NOT active phages/living bacteria. It will count the dead/inactive ones just the same. Adding in culture-based methods (culture the bacteria, do plaque assays on the serum, etc) is helpful and needed to start getting a sense of who’s alive or dead. Still, qPCR can give you a sense of how many copies of the DNA of the organism you’re trying to “watch”, and how that’s changing over time. Still very interesting!
⛔ Another caveat: this is not a qPCR tutorial, it’s just an overview, written by a beginner, to get you thinking about whether this method is something you’d like to try, and help you wrap your head around the steps if you’re brand new to it like I was a few months ago. There are many much better qPCR resources out there, and I’m glossing over a lot here!
Basic intro to qPCR
qPCR is where you do PCR in the presence of a DNA-binding fluorescent dye (like SYBR green). Throughout the reaction (in real time), the machine detects the fluorescence, which goes up as more and more DNA is created with each cycle of the ~40-cycle reaction. The whole point is to quantify how much target DNA was there to begin with.
If there is a lot of starting DNA in your sample, it won’t take too many cycles for the machine to detect the fluorescence. If there’s only a little starting DNA in your sample, it will take many cycles to create a detectable amount.
For each sample we run, the machine will record how many cycles out of ~40 it took to create a detectable* amount of fluorescence. This is the Cq (aka Ct, or ”cycle threshold”) number.
By creating a series of “standards”, where we do PCR on a bunch of different known quantities of our target, we can get a Cq number for each. We can use those numbers to generate a mathematical relationship between Cq and starting quantity, and then use that relationship to convert Cq of our samples into a more meaningful unit, like PFU/mL or number of copies of phage X DNA per mL.
*Detectable: not literally what is detectable, but detectable above noise
Preparing for qPCR on patient serum samples
Doing qPCR to detect phage or bacteria in patient serum samples is essentially the same as doing it for any other type of target/sample, with just a few tweaks. Essentially, you’ll design primers, create standards (spiked into your sample matrix type, ie. serum), extract DNA from standards and patient samples, set up PCR reactions, run the machine, and get your results.
(See Khatami et al. 2021, who tracked a phage therapy case using this method, and Khatami et al. 2022, which describes the STAMP Study which incorporates this method)
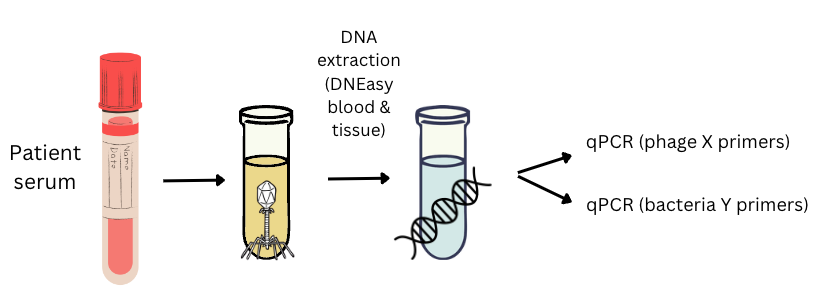
From a high-level view, this is roughly what you’ll need:
- Primers designed to amplify your phage(s)
- Primers designed to amplify your target bacteria
- Samples from the patient getting phage therapy (probably blood or serum samples, but you could do this in urine, fecal samples, or really any sample type)
- An aliquot (of approximately known concentration) of each phage or bacteria you’re trying to detect
- (Optional): a sample of healthy volunteer serum/blood (or whatever matrix your samples are in)
- DNA extraction kit/protocol that reliably extracts DNA from your sample type (if it’s serum, the DNA extraction kit should be known to work on serum)
- A way to check quantity and quality of DNA (ie. Nanodrop)
- qPCR kit/protocol, including reagents (e.g. fluorescent DNA-binding dye like SYBR, nuclease-free water)
- A qPCR machine
Why do we need standards again?
Standards are a big thing to wrap your head around with qPCR; they pretty much underpin the entire assay. You might have heard ‘you need a standard curve!’. But why do we need it again?
Let’s work backwards from what the qPCR machine actually gives us: a Cq number. Let’s say you ran your samples and got your Cq numbers. You might excitedly say ‘after the patient’s first phage dose, there was Cq = 17 phages in there! Isn’t that cool?’ (No… I don’t know what Cq = 17 means). Does anyone?
No. Without standards, Cq numbers don’t mean anything. The only way to know what Cq = 17 means is to run a series of standards (a bunch of known quantities of the thing you’re trying to measure, spanning the range you care about), graph them, and use the equation of that line to convert your Cq values into values like PFU/mL.
Example: if you think you might have anywhere from 0 to 10^7 pfu/ml of a phage in your patient’s blood samples, you might choose to create standards that span that whole range, ie. 0, 10^1, 10^2, 10^3, 10^4, 10^5, 10^6, 10^7 pfu/ml of that phage (8 standards). Then, you’d run those 8 standards on your qPCR machine, and you’d get Cq values for each of them. Maybe the 10^7 gave you 17, and the 10^6 gave you 20, and the 10^5 gave you 23… and so on. You’d collect all that, and use it to generate your standard curve and equation of the line, which you could then use to convert your unknown Cq values into PFU/mL values (we won’t get into the math today; perhaps in a future post).
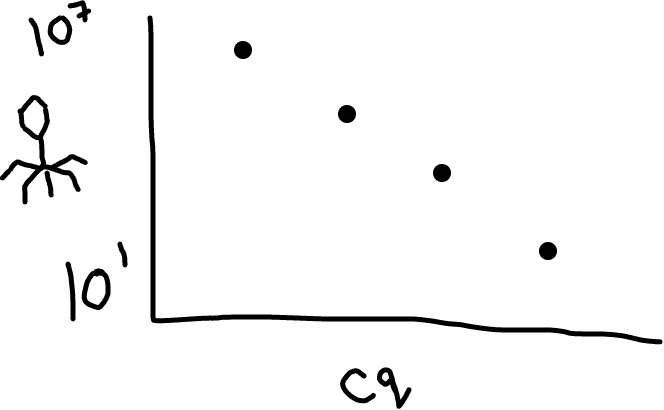 Fig 2: Higher amounts of phage in your sample will lead to a lower Cq value (it’s an inverse relationship).
Fig 2: Higher amounts of phage in your sample will lead to a lower Cq value (it’s an inverse relationship).
Wait, how do I actually make the standards?
This is the fun part! If you’re trying to measure phage X, you’ll need an aliquot of phage X at a known concentration. If you’re trying to measure bacteria Y, you’ll need an aliquot of bacteria Y at a known concentration.
To get a known concentration, you’ll need to obtain a fresh stock of each organism you’re detecting (whether phage or bacteria) and do a titre (plate for PFUs or CFUs). If you’re using a phage, you might start around 10^8 PFU/mL (or higher, if you’re expecting higher numbers in your patient samples). For a bacterium, take a few colonies and dip them into broth or buffer, and use a Macfarland reader to set the suspension to around 0.5 — this will give you roughly 10^8 CFU/mL (will change for different species, but at this stage it’s ok if it’s not exact).
Use these estimated counts to start with, but also plate out to get real counts (these real counts are what you’ll actually use to make your standard curves; details later). For now, move right on to the next step while those plates are incubating.
For each organism you’re detecting, you’ll want to make a dilution series so you end up with vials of each concentration in your range. You may want to make that dilution series in a matrix that approximates what your patient samples are in. (Example: you could make your dilution series of phage in human serum if that’s what your patient samples are in).
Why might you do this? Well, the next step is DNA extraction, which might not work that well in presence of serum (or feces/urine/blood for that matter). So if we’re taking patient samples from one of those mucky environments, we should probably make sure our standards are taken from the same thing. That way if DNA extraction is only half as efficient in presence of serum (ie. only half the DNA in the serum is actually ‘captured’ during the DNA extraction step), at least the standards will reflect that as well, so this error will ultimately be cancelled out.
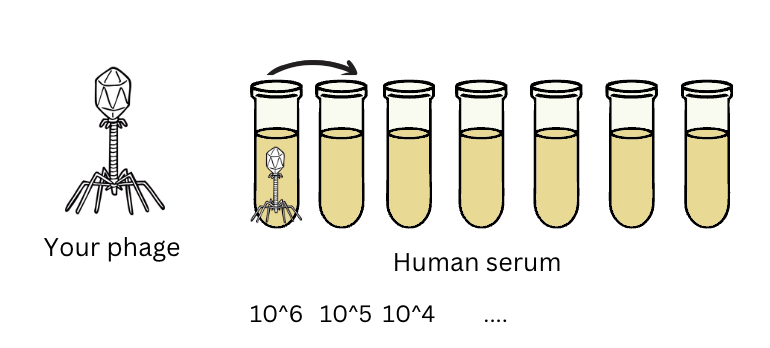
Fig 3: If you’re extracting phage from serum, it’s a good idea to spike your standards into serum too, to make sure your qPCR assay works to quantify your phage even in the presence of all the random things floating around in serum (these might interfere with DNA extraction or with PCR).
DNA extractions
This part is easy — there are so many kits for DNA extraction, and if you don’t have a kit, you can just follow protocols online which use generally available materials like detergent, ethanol, buffer, salts. The important thing is that all your samples and all your standards are DNA-extracted in the same way!
You’ll want to do DNA extractions for each patient sample and each standard. I’ve recently tested out replicate extractions from the same patient serum sample, and there wasn’t too much of a spread. I think it’s a good idea to get a sense of variability at each step of any protocol if you can. That said, if resources are constrained (as they usually are), it’s also not unreasonable to assume that the DNA extraction kit/protocol you’re using, especially a commercial kit, will perform roughly consistently each time for the same sample type, and roughly the same for standards as samples (assuming we’ve spiked our standards into the relevant matrix). So you can probably get away with doing one DNA extraction per patient sample.
For standards, it’s nice to do a duplicate dilution series (to capture variation from pipetting error), and do a DNA extraction from each.
Information to gather before each qPCR run
At this stage it’s nice to start a spreadsheet, and fill it in with basic info about your samples, standards, and reaction conditions as you set up for each qPCR run (this is just the basics; see MIQE guidelines for more comprehensive list and reasons for what you should collect before publishing qPCR data)
Standards
1. Name/standard ID (e.g. Std1)
2. Description of standard (e.g. DNA extracted from phage X at 10^7 pfu/mL in serum; undiluted)
3. Amount of DNA (in ng/uL)
4. DNA quality (260/280; 260/230)
Samples
1. Name/sample ID (e.g. 2A)
2. Description of sample (e.g. day 2, prior to phage administration)
3. Amount of DNA (in ng/uL)
4. DNA quality (260/280; 260/230)
PCR reaction ingredients
1. Which primers? How much of each?
2. Which reagents?
3. How much template DNA?
PCR conditions
1. How many minutes at each temperature? (If following a master mix kit, they may specify ideal PCR conditions to start with; I usually start with those when in doubt)
Does DNA quantity/quality matter?
At this stage, you’ll wind up with DNA, and it’s a good idea to quantify it. Why? Because if your samples have really high amounts of DNA, you might need to dilute it before adding to your qPCR reaction (if you overload your reaction, it might not work/you might get a garbage reading). If they’re really low, you will want to use it undiluted (if it’s too low, the primers won’t be able to find their targets and you’ll get no PCR amplification/no reading). Note: if you do make any dilutions, make a note! These will need to be taken into account later since this is a quantitative assay.
You’ll also want to check out quality of your DNA using a nanodrop or other method. If your DNA is of really poor quality, you might want to tweak your DNA extraction protocol. That said, low concentrations of DNA are usually going to give poor quality readings, just based on the limitations of the machines.
While good quality numbers would be great, you’re probably not going to be able to be too picky if you’re trying to get DNA out of patient blood samples… it’s probably not going to be a lot, and thus it’s not going to give you a beautiful DNA curve on the nanodrop. My rule of thumb is generally: when in doubt, make a note of the quantity/quality, and move on with your PCR even if the numbers don’t look great.
(This is an area where you really shouldn’t take my word for it; I’m still figuring this out. Please read up on qPCR from the experts to understand to what extent DNA quality matters at this stage!).
Primers: designing the right ones
Primer design is a whole art in itself, which I won’t get into in this post. The main thing is, if your patient was treated with multiple phages, you’ll want to make sure you have primers targeting each phage, and that your primers for each phage do not amplify any of the other phages that will be present.
You can pick any gene to target in the phage; maybe the DNA polymerase, or the primase, whatever you like.
To amplify your target bacteria, a nice shortcut is to look up primers used in the literature for that species, e.g. this paper describes gadE primers that work on most E. coli.
You can check if the primers “should” amplify your target through online tools. You can also check if they “should not” amplify the other phages in your mix (and that they won’t amplify human DNA, which is important to check given we’re amplifying from patient samples). But you won’t really know if the primers work, or if they’re cross-reactive with the other phages, until you try them.
Also, you may have heard of multiplexing. This is not really necessary; you don’t need to multiplex, ie. have all your PCRs amplifying in the same tube. You can just do separate PCRs on each sample with each separate set of primers.
Mapping out the run
Your kit or protocol for qPCR will specify how much of each reagent should go in a reaction. To make a master mix, you’ll need to multiply those values by the total number of reactions you’ll be doing. How many is that?
This is the time to plan out your run. How many samples do you have? How many standards do you have? What about controls?
Let’s do this as an example. Say you have one phage to detect, and you have 14 patient samples. You have made standards (8 dilutions of your target phage, spiked into serum; triplicate PCR reactions). One of these 8 is actually a negative control (zero phage; ie. serum only). You also want a crossover control for each primer set + each non-target phage. (e.g. if you have given the patient 2 phages, each primer set needs to be checked for cross-reactivity with the other phage, so you’ll need 2 crossover controls; also run these in triplicate).
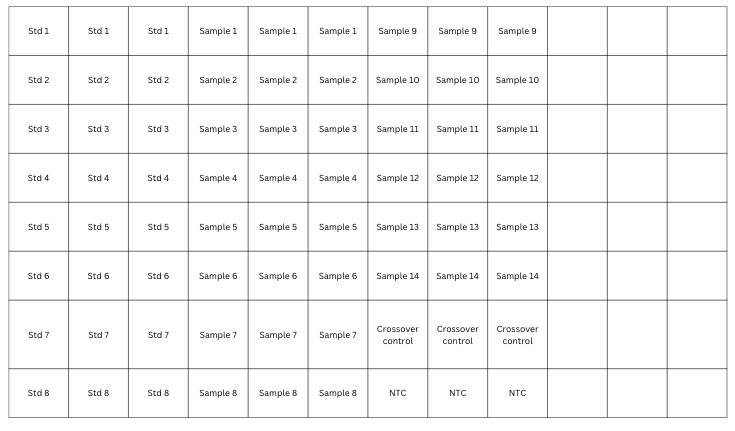
Fig 4: 96-well plate map showing a sample qPCR run; this one has 8 standards, 14 samples, and 2 controls; each run in triplicate. Note this is for one primer set.
Now that you’ve mapped out your standards, samples and controls, add up how many total PCR reactions you’ll be setting up. From the above, I have 72. For my master mix, I’ll want to have 72+ a bit extra for pipetting error; let’s say we round up to 75.
Running the qPCR
From here, setting up qPCR is the same as setting up regular PCR; make the master mix, distribute into wells, add the template DNA, load samples into the machine, set the machine to your desired temperature cycling conditions, press start, and come back in a couple hours.
The nice thing about qPCR is that the samples don’t need to be run on a gel! The computer will detect how much fluorescent dye is generated (a proxy for how much PCR product is produced) over time, for each well.
Whatever machine you use will likely have software along with it. It’ll generate graphs for each of your samples. To do this, it’ll ask you for the names of what’s in your wells, and for the starting quantity of each of your standards (10^7, 10^6, and so on). Fill these out based on real values for your phage/bacteria (the counts you got from plating your “known quantity” on the day you made your standards).
Stay tuned for part II!
In part II we’ll go into what we should be looking for as we analyze our results. Did our primers work? Was our PCR reaction efficient? Is our standard curve useable? Assuming all these answers are yes, it’s finally time to take a look at our samples: how much phage / bacteria do we see in our patient serum samples?
Further reading
- BiteSizeBio qPCR guide
- Taylor, S. C. et al (2019). The ultimate qPCR experiment: producing publication quality, reproducible data the first time. Trends in Biotechnology, 37(7), 761-774.
- Paper using qPCR to follow a phage (and Pseudomonas target) in a patient’s serum samples during phage therapy (I’ve been following the methods used here): Khatami, A. et al. (2021). Bacterial lysis, autophagy and innate immune responses during adjunctive phage therapy in a child. EMBO Molecular Medicine, 13(9), e13936.
- STAMP Study, which includes qPCR to track phages and bacteria in serum: Khatami, A. et al. (2022). Standardised treatment and monitoring protocol to assess safety and tolerability of bacteriophage therapy for adult and paediatric patients (STAMP study): protocol for an open-label, single-arm trial. BMJ open, 12(12), e065401.
- Paper doing qPCR on 5 phages and comparing with plaque assays: Duyvejonck, H. et al. (2019). Development of a qPCR platform for quantification of the five bacteriophages within bacteriophage cocktail 2 (BFC2). Scientific reports, 9(1), 13893.
- Paper using qPCR to follow a bunch of phages in different cocktails vitro: Wandro, S. et al. (2022). Phage Cocktails Constrain the Growth of Enterococcus. Msystems, 7(4), e00019-22.